Hereditární amyloidóza
Vznik hereditární amyloidózy je charakterizován mutací neboli záměnou genetické informace (DNA) v genu, který je zodpovědný za tvorbu některého z proteinů, který může dát vzniknout amyloidu. V molekulách DNA (deoxyribonukleové kyseliny) se uchovává genetická informace živých organismů a díky ní dochází k přenosu dědičných znaků na potomstvo. Zároveň DNA slouží jako jakási předloha pro vznik proteinů, které jsou životně důležité stavební prvky organismů a významné látky pro funkci metabolismu.
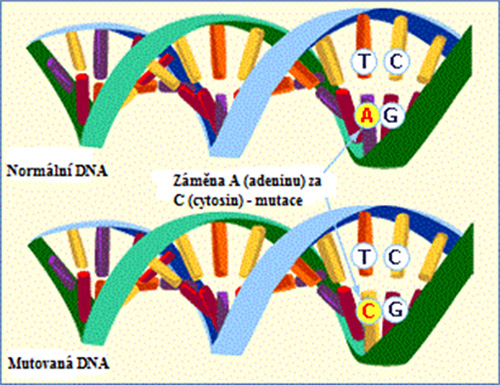
Obrázek 1: Záměna na úrovni DNA, která je zodpovědná za tvorbu amyloidu. Ke vzniku amyloidu stačí záměna jedné báze (např. A za C neboli dojde ke změně adeninu za cytosin). Tento stav označujeme jako tzv. mutaci v genu.1
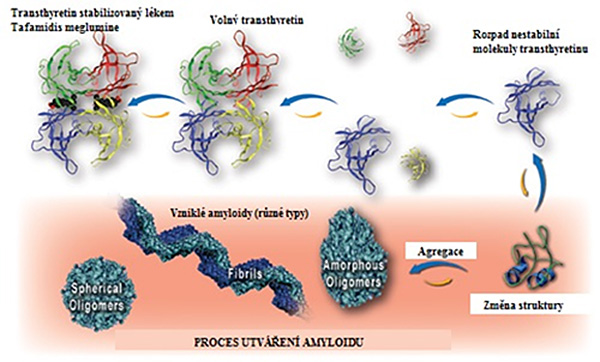
Obrázek 2: Popis procesu utváření amyloidu u transthyretinové amyloidózy. Nestabilní transthyretin se rozpadá z homotetrameru (tzn. že se skládá ze čtyř identických podjednotek, v obrázku odlišeno 4 barvami) na monomery (jednotlivé jednotky např. modrá). Tyto jednotky pak prochází změnami struktury, díky kterým jsou odolné vůči odbourávání a snadněji podléhají procesu, který vede ke vzniku amyloidu. Výsledkem je vznik amyloidů, které mohou různý tvar.2
Tato záměna způsobí, že nově vznikající protein bude mít pozměněnou strukturu a tím i pozměněné vlastnosti. Obecně k těmto vlastnostem patří nestabilita, změněné prostorové uspořádání proteinu, neschopnost odbourat se a jednodušší utváření depozit amyloidu.
V dnešní době se počet genů, v nichž mutace může způsobit hereditární amyloidózu, udává kolem 22. Mezi geny, které bývají mutovány nejčastěji, se řadí TTR (gen kódující vznik proteinu transthyretinu) a APO A-I (gen kódující vznik proteinu apolipoprotein A1). V ostatních genech jsou mutace méně časté nebo raritní.
Transthyretinová hereditární amyloidóza (ATTR)
Transthyretinová hereditární amyloidóza je charakterizována ukládáním depozit proteinu transthyretinu. Transthyretin (prealbumin) v lidském těle plní funkci transportního hormonu (látky, které v organismu hrají roli chemických poslů od jedné buňky k druhé). V tkáních se může vyskytnout nejen jeho mutovaná forma (depozice neboli ukládání je způsobeno mutací v genu, tedy chyba nastává při vzniku proteinu, vzniká mutovaný protein), ale také jeho nativní forma (depozice je způsobena chybou v procesu odbourávání proteinu, mutace se v genu nenachází, tedy chyba je ve špatném odbourávání normálně vzniklého proteinu). U hereditární formy je v dnešní době známo přibližně 120 mutací, které mohou způsobit změnu v proteinu. Mutace v genu se mohou v rámci rodiny dědit na potomky, a to nejčastěji tzv. autosomálně dominantně.
Klinická manifestace (projev nemoci) transthyretinové amyloidózy závisí na typu (druhu záměny) mutace. Nejčastějším klinickým projevem je tzv. familiární amyloidní polyneuropatie (FAP). Familiární amyloidní polyneuropatii řadíme mezi život ohrožující multiorgánové onemocnění, které je způsobeno depozicí amyloidu v tkáních, nejvíce v periferních nervech. Následkem depozice v nervech je vznik lézí, klinická manifestace může být variabilní. Nemoc se ze začátku projevuje ztrátou citlivosti v dolních končetinách, pokračuje ztrátou na váze a celkovou sešlostí. Častými příznaky můžou být úzkost, závrať, únava, mravenčení, brnění nebo bolest svalů končetin. Jiným klinickým projevem může být familiární amyloidní kardiomyopatie (FAC), kde k depozici amyloidu dochází mezi buňkami srdce. Příznaky kardiomyopatie můžou být dušnost, únava, nepravidelnost srdečního rytmu, v pozdních fázích pak srdeční nedostatečnost, vedoucí až k smrti. Obecně je familiární amyloidní kardiomyopatie spojována s horší prognózou než familiární amyloidní polyneuropatie. Průměrný věk manifestace jak FAP, tak FAC je u mužů i žen kolem 40–50 roku života. Přežití v případě neléčené nemoci je udáváno v průměru 10 let. Léčebné možnosti jsou neustále ve fázi vývoje nových preparátů pro zkvalitnění a prodloužení života pacientů. V současné době je pro léčbu FAP registrováno léčivo Tafamidis meglumine (Pfizer), které působí jako stabilizátor transthyretinu a tím blokuje vznik nestabilních molekul transthyretinu, které jsou podmínkou pro vytváření amyloidu. Další preparáty jsou ve fázi klinického testování. Jediným možným řešením pro úplné vyléčení je transplantace postiženého orgánu (srdce v případě FAC) nebo transplantace orgánu, který je hlavním producentem mutovaného proteinu (játra v případě FAP), u některých pacientů není výjimkou transplantace jak srdce, tak jater. Transplantace však není vhodnou metodou léčby pro pacienty se všemi druhy mutací, tedy i její možnosti jsou zatím omezené.
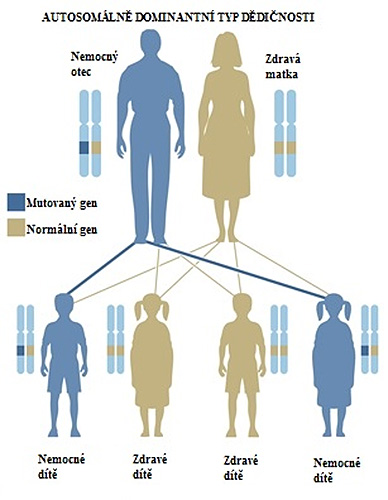 Obrázek 3. Schéma autozomálně dominantní dědičnosti. V tomto případě vycházíme z předpokladu, že každý z nás má ve své buňce každý zděděný gen ve dvou kopiích. Jednu kopii genu dědíme od otce a druhou od matky. Ze schématu vyplývá, že pokud potomek zdědí jen jeden mutovaný gen (v tomto případě od otce), nemoc se u něho projeví, i když druhý gen, který zdědil od matky je zdravý. U tohoto schématu teda záleží na tom, který ze dvou genů od otce zdědíme, zdali ten mutovaný, nebo zdravý, přičemž šance na zdědění jakéhokoliv ze dvou genů je přesně 50 %. Tedy pravděpodobnost, že potomek rodiče, který trpí transthyretinovou hereditární amyloidózou, ji zdědí, je přesně 50 %.3
Obrázek 3. Schéma autozomálně dominantní dědičnosti. V tomto případě vycházíme z předpokladu, že každý z nás má ve své buňce každý zděděný gen ve dvou kopiích. Jednu kopii genu dědíme od otce a druhou od matky. Ze schématu vyplývá, že pokud potomek zdědí jen jeden mutovaný gen (v tomto případě od otce), nemoc se u něho projeví, i když druhý gen, který zdědil od matky je zdravý. U tohoto schématu teda záleží na tom, který ze dvou genů od otce zdědíme, zdali ten mutovaný, nebo zdravý, přičemž šance na zdědění jakéhokoliv ze dvou genů je přesně 50 %. Tedy pravděpodobnost, že potomek rodiče, který trpí transthyretinovou hereditární amyloidózou, ji zdědí, je přesně 50 %.3
Apolipoprotein A1 amyloidóza (AApoA I)
Hereditární amyloidóza spojená s mutací v genu pro apolipoprotein A-II patří mezi autozomálně dědičná onemocnění. Protein apolipoprotein A-II se vyskytuje se v plasmě a jeho hlavní funkcí je transport cholesterolu k buňkám. Amyloidní depozita, u osob postižených tímto druhem onemocnění, se nachází primárně v ledvinách, kde mohou způsobit poškození až selhání ledvin, ale mohou se vyskytovat i v jiných orgánech např. srdci, slezině. Svými projevy se může podobat amyloidóze AFib (amyloidóza způsobená mutací v genu pro fibrinogen) nebo ALys (amyloidóza způsobená mutací v genu pro lysozym). Prozatím jediným dostupným způsobem léčby je dialýza následovaná transplantací ledvin.
Fibrinogen (AFib)
Mutace v genu pro protein fibrinogen jsou spojovány se vznikem hereditární amyloidózy označované jako AFib. Jedná se o autozomálně dominantní onemocnění. AFib je nejčastějším typem hereditární amyloidózy spojeným s poškozením ledvin v Evropě. Fibrinogen je protein vyskytující se v plazmě a hraje důležitou roli při procesu srážení krve. Druh mutace v genu může mít vliv na nástup nemoci, určité druhy mutací způsobí projevy nemoci již v dětském věku, jiné naopak až v středním věku. Tento typ amyloidózy může způsobit postižení především ledvin, dále pak srdce, nervů (autonomní nervový systém) popř. jiných orgánů (játra, slezina, trávící trakt). Léčba AFib je primárně transplantační, dle pokročilosti postižení ledvin lze zvážit kombinovanou transplantaci ledvin a jater.
Gelsolin (AGel)
Gelsolinová amyloidóza (AGel) známá také pod názvem familiální amyloidóza finského typu je dědičné onemocnění s autozomálně dominantním typem dědičnosti, které je způsobené mutací v genu pro protein gelsolin. AGel je onemocnění vyskytující se s vyšší frekvencí ve Finsku, kde také bylo poprvé popsáno, v jiných zemích je však vzácnější. Gelsolin je protein plnící důležitou funkci především v buněčné biologii (pohyb buněk aj.), ovlivňuje také viskozitu krve a odpověď organismu na záněty. Zajímavostí je gelsolinu je, že jedna z mutací byla popsána jako typická pro českou populaci. Depozita amyloidu gelsolinu bývají nacházena v srdci, ledvinách, nervech a dalších útrobních orgánech. Gelsolin mimo jiné řadíme mezi proteiny, jejichž mutace může způsobit tzv. hereditární formou mozkové amyloidní antipatie (tzn. nezánětlivé onemocnění cév). Částečně se může podílet na vzniku Alzheimerovy choroby. AGel se může vyskytnout také jako ojedinělá forma a to v kombinaci s AA amyloidózou, mnohočetným myelomem, AL amyloidózou nebo DM 2. typu (diabetes mellitus 2. typu, cukrovka 2. typu). Obvykle AGel není spojena s kratším přežitím.
Lysozym (ALys)
ALys je označení pro formu hereditární systémové amyloidózy s autozomálně dominantním typem dědičnosti způsobená výskytem mutace v genu pro protein lysozym. Ve srovnání s ATTR se jedná o vzácný typ hereditární amyloidózy. Protein lysozym můžeme najít ve slinách, slzách, plasmě nebo mateřském mléce, jeho role v organismu však stále není přesně objasněna. Projev této nemoci je variabilní a závisí na věku a postiženém orgánu, zároveň se může lišit u osob se stejným typem mutace. Pokud se však mutace dědí v rámci rodiny, příznaky bývají podobné. ALys bývá často mylně zaměňována za AL amyloidózu. Společným rysem ALys je postižení ledvin, míra afekce dalších jiných orgánů je variabilní, nebývá postiženo srdce, ale jen zřídka bývá postižený nervový systém (periferní). Orgánové poškození při ALys zahrnuje také postižení trávícího traktu, jater, sleziny, slinných žláz. Specifická terapie není k dispozici, ani transplantace jater není přínosem, jelikož lysozym vzniká na mnoha místech organismu.
Cystatin C (ACys)
Hereditární amyloidóza spojená s mutací v genu pro cystatin C neboli hereditární angiopatie islandského typu je onemocnění s autozomálně dominantní dědičností. Protein cystatin C je produkován mnoha typy buněk v organismu a stanovení jeho hladiny se používá jako indikátor funkce ledvin. Depozice vzniklého amyloidu nastává především v mozku (mozkových plenách, mozkovém kmeni, mozečku aj.). Depozita lze také najít v lymfatických orgánech, kůži, slinných žlázách nebo ve varlatech. Jedná se o onemocnění často s fatálním průběhem, časným krvácením do mozku popř. nástupem demence, průměrný věk nástupu nemoci je 30 let. Cystatin C může hrát roli při vzniku jiných druhů amyloidóz a může přispívat ke zvýšenému riziku vzniku Alzheimerovy choroby. Pro ACys neexistuje specifická terapie. Předpokládá se, že zvýšená teplota může přispívat ke vzniku amyloidů, proto se doporučuje předcházet a časně léčit horečnaté stavy
1 Illustration used with permission from Aetna InteliHealth. Medical content reviewed by the Faculty of Harvard Medical School. Copyright 1996-2014.
2 Zdroj: BULAWA, C. E., S. CONNELLY, M. DEVIT, L. WANG, C. WEIGEL, J. A. FLEMING, J. PACKMAN, E. T. POWERS, R. L. WISEMAN, T. R. FOSS, I. A. WILSON, J. W. KELLY a R. LABAUDINIERE. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proceedings of the National Academy of Sciences. 2012, vol. 109, issue 24, s. 9629-9634. DOI: 10.1073/pnas.1121005109.
3 Zdroj: http://ghr.nlm.nih.gov/handbook/illustrations/autodominant.